Targeted protein degradation has emerged as a powerful therapeutic strategy, but most approaches only reach proteins inside cells. Roughly 40% of the human proteome consists of extracellular and membrane proteins that lie beyond the reach of conventional degraders. Lysosome-targeting chimeras address this gap by hijacking the lysosomal pathway, recruiting cell-surface receptors that shuttle bound cargo to lysosomes for destruction. The first-generation LYTACs used polymeric glycopeptides to engage the cation-independent mannose-6-phosphate receptor, but their heterogeneous composition complicated therapeutic development.
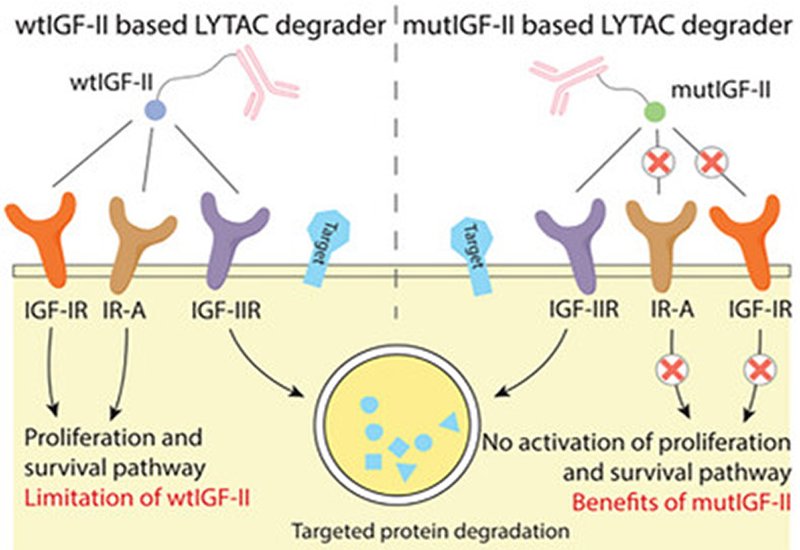
Researchers in the Tang Group at the University of Wisconsin-Madison, in collaboration with colleagues at NovoNordisk, published in Advanced Science, have engineered an insulin-like growth factor II mutant that offers a cleaner solution. The mannose-6-phosphate receptor is identical to the type II IGF receptor, making native IGF-II an attractive alternative ligand. The problem is that wild-type IGF-II also activates the type I IGF receptor and insulin receptor isoform A, triggering mitogenic signaling that promotes cell growth and survival. For a degrader platform, such off-target activity poses unacceptable safety risks.
The team combined two previously reported mutations, Del1-7 and Y27L, into a single IGF-II variant they call mutIGF-II. Surface plasmon resonance measurements confirmed that mutIGF-II binds the type II receptor with nanomolar affinity comparable to wild-type IGF-II. Binding to the type I receptor and insulin receptor isoform A was undetectable. Competition assays reinforced this selectivity: a five-fold molar excess of mutIGF-II abolished wild-type IGF-II binding to the type II receptor while leaving interactions with the other receptors intact. In cellular assays measuring AKT phosphorylation, a readout of mitogenic signaling, mutIGF-II showed negligible activity even in cells overexpressing the type I receptor or insulin receptor.
Conjugating mutIGF-II to biotin created a degrader targeting fluorescent NeutrAvidin as a model cargo. The construct induced dose-dependent internalization and lysosomal trafficking in HepG2 cells, with confocal microscopy confirming colocalization with lysosomes. The characteristic bell-shaped dose-response curve indicated productive ternary complex formation between receptor, degrader, and cargo. Extending this approach to therapeutically relevant targets, the researchers conjugated mutIGF-II to antibodies against EGFR, PD-L1, and Her2. Treatment with 10 nM of the EGFR degrader induced 70 to 80% receptor degradation in Huh7 cells and suppressed downstream ERK phosphorylation more effectively than the parent antibody cetuximab alone. The PD-L1 degrader achieved 60 to 70% degradation in MDA-MB-231 cells at 1 nM.
Comparing mutIGF-II degraders to glycopeptide-based constructs revealed striking differences in potency. The glycopeptide ligand bound the type II receptor with roughly 300 nM affinity versus 7 nM for mutIGF-II. This 40-fold difference translated directly into functional outcomes: 10 nM mutIGF-II degrader achieved 80 to 90% PD-L1 degradation in U87 cells while the glycopeptide degrader at the same concentration had no effect. Knockdown experiments confirmed that mutIGF-II degrader activity depended entirely on type II receptor expression, whereas wild-type IGF-II degraders retained partial activity through alternative receptor engagement.
The platform extends beyond chemical conjugation. A genetically encoded fusion protein linking mutIGF-II to a PD-L1-targeting single-chain antibody fragment was secreted from transfected cells and induced target degradation in neighboring untransfected cells. In three-dimensional tumor spheroids, mutIGF-II constructs penetrated deep into the spheroid interior rather than accumulating only at the periphery. The engineered selectivity eliminates mitogenic liabilities while preserving potent lysosomal targeting, establishing mutIGF-II as a foundation for safer extracellular protein degraders.