Selective chemical modification at a single, predictable site remains a persistent challenge in peptide and protein chemistry. N-terminal cysteine offers an attractive handle because its 1,2-aminothiol motif is rare in native proteomes, since translation begins from methionine, and can be targeted with high selectivity. Most reagents developed for this handle, from 2-cyanobenzothiazole to cyclopropenone derivatives, share one outcome. They drive a tandem addition that consumes both the amine and the thiol to form an inert heterocycle. The ligation site becomes a dead end, blocking further diversification and closing off higher-order architectures such as bicyclic peptides.
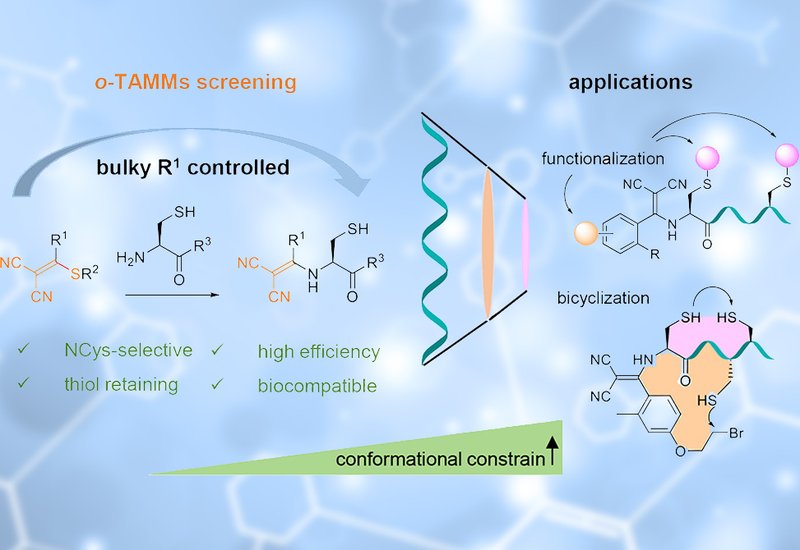
Researchers at Shenzhen Bay Laboratory, Xiamen University, and Taiwan's National Yang Ming Chiao Tung University, published in the Journal of the American Chemical Society, traced this thiol loss to a thermodynamic preference built into the reagent scaffold. In the reaction of 2-((alkylthio)(aryl/alkyl)methylene)malononitriles, TAMMs, with 1,2-aminothiols, a thiol-retaining enamine forms first but converts to a thermodynamically favored dihydrothiazole, sequestering the thiol. The team reasoned that steric congestion at the β-position of the TAMM aryl ring would raise the barrier for that cyclization and hold the reaction at the enamine stage. They prepared a series of ortho-substituted TAMMs, o-TAMMs, and tested them with N-terminal cysteine peptides in bicarbonate buffer.
The hypothesis held. Ortho-methyl TAMM 1a reacted with the model peptide H-CGGGKGW-OH to give the thiol-containing enamine as the sole detectable product, with no conversion to dihydrothiazole over 48 hours. Bulky ortho substituents favored enamine retention, while small substituents such as fluoro and methoxy allowed cyclization to proceed. Calculations placed the steric effect on a quantitative footing, raising the effective barrier for malononitrile elimination from 21.8 to 26.3 kcal/mol. The retained thiol stood ready for downstream chemistry. Sequential maleimide capping followed by copper-catalyzed azide-alkyne cycloaddition gave dual-functionalized peptide conjugates in a one-pot workflow, and the same approach carried over to proteins bearing an N-terminal cysteine, including the affibody zHER2 and SUMO.
The retained thiol also opened a route to compact bicyclic peptides. The team fitted an o-TAMM with a para-substituted alkyl bromide to build the bifunctional cross-linker 1q. Reacted with CXmCXnC peptides, 1q forms the enamine thioether at the N-terminal cysteine, the alkyl bromide then substitutes chemoselectively at an internal cysteine to close a thioether ring, and oxidation joins the remaining two thiols into a disulfide ring. Phage display against KEAP1, the negative regulator of the NRF2 antioxidant pathway, yielded binders after three rounds. The lead sequence A3-4 gave bicyclic isomers BCP-a and BCP-b with dissociation constants of 312 and 108 nM by surface plasmon resonance, against 4160 nM for a fully linearized analogue. The bicyclic topology delivered the strongest and most consistent affinity, while single-ring controls bound more weakly.
The o-TAMM platform turns a fleeting intermediate into an isolable, versatile product under mild aqueous conditions. The thioether-disulfide bicyclic scaffold it produces resists glutathione reduction better than a simple disulfide, and its disulfide converts to a redox-stable thioacetal without loss of KEAP1 affinity. The reagents react about 20-fold slower than their para-substituted counterparts, though electron-withdrawing thiol substituents such as trifluoroethanethiol offer a faster variant. The authors point to switchable chimeric antigen receptor T-cell systems, where TAMM condensation in vivo could time the assembly of a recognition module.