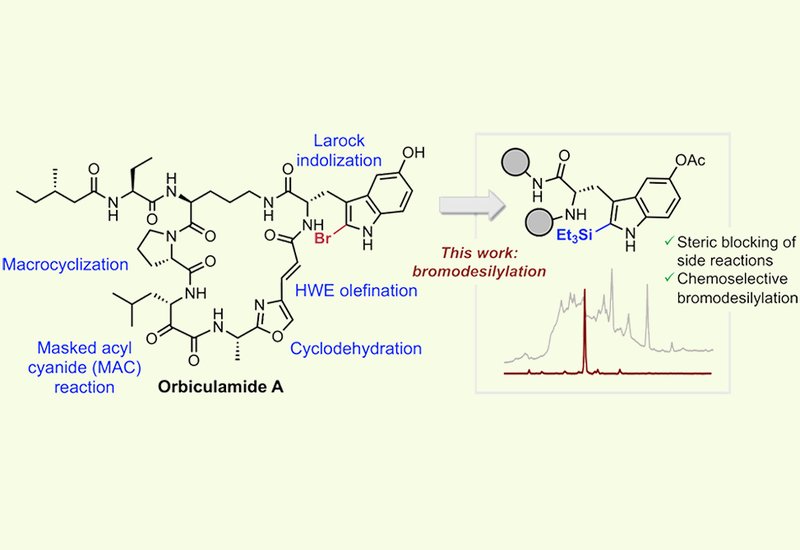
Marine macrocyclic peptides have long captured the imagination of synthetic chemists and pharmacologists alike, but converting their structural complexity into tractable synthetic targets remains a formidable challenge. Orbiculamide A, first isolated in 1991 from a Theonella sponge, is a cytotoxic macrocycle with antimitotic and antileukemic activity. Its resistance to total synthesis over more than three decades stems from three compounding structural features: an α-keto-amide, a trans-olefin bridging oxazole, and, most critically, a 2-bromo-5-hydroxytryptophan, 2-Br-5HTrp, residue. Electrophilic bromination of tryptophan and tryptophan-containing peptides is notoriously plagued by oxindole formation, spirolactone-mediated peptide hydrolysis, and 3a-bromopyrroloindoline ring closure, leaving no reliable late-stage route to place the C2-bromo substituent within a complex macrocycle.
Researchers in the Perrin Group at the University of British Columbia, published in Organic Letters, addressed this long-standing challenge by designing a retrosynthetic strategy built around a chemoselective, late-stage bromodesilylation. The key insight was that a bulky triethylsilyl, TES, group at C2 of the indole, installed via a Larock indolization of a macrocyclic alkyne-bearing precursor, could simultaneously block unwanted side reactions and serve as a masked handle for subsequent electrophilic bromination. Solid-phase peptide synthesis, SPPS, provided the convergent backbone, while a masked acyl cyanide, MAC, reaction furnished the theonalanine building block bearing the α-hydroxy-amide precursor to the α-keto-amide.
Optimization of the bromodesilylation on a model substrate revealed that N-bromosuccinimide, NBS, in acetonitrile delivered 96% conversion to the desired 2-bromo product within two hours at room temperature, outperforming all other electrophilic brominating agents and solvent systems tested. Isolation of a C3-brominated indolenine intermediate as a single diastereomer, confirmed by diagnostic Nα-H:Cα-H NMR correlations, suggests a mechanism involving C3 bromination followed by a 1,2-bromine shift with concomitant desilylation rather than direct ipso-substitution. X-ray crystallography definitively confirmed the structure of the desilylated product. Direct bromination of a tryptophan-containing precursor lacking the TES group gave less than 20% conversion under identical conditions and produced extensive side products, including the 3a-bromopyrroloindoline adduct, underscoring the indispensability of the silyl-protecting strategy.
The sequence that delivered orbiculamide A required careful choreography of oxidation and bromination steps. After Larock indolization of the macrocycle, selective removal of the tert-butyldimethylsilyl, TBS, group on the α-hydroxy-amide side chain left the C2-TES-indole intact. Dess–Martin periodinane oxidation then converted the free alcohol to the α-keto-amide in 72% yield. Crucially, the TES group resisted hypervalent iodine-induced indole oxidation, enabling this transformation to proceed without compromising the indole. Bromodesilylation of the oxidized intermediate with NBS in acetonitrile at 60 °C gave the 2-Br-5-O-acetyl-Trp macrocycle in greater than 80% yield, and mild base-mediated acetyl deprotection with methanol furnished orbiculamide A with greater than 90% conversion by LCMS and isolated yields exceeding 60%. The spectroscopic data matched those of the natural product, though the optical rotation of the synthetic material differed from the originally reported value, a discrepancy consistent with known complexities in natural product structure elucidation.
Beyond providing the first total synthesis of orbiculamide A, this work establishes a generalizable blueprint for late-stage indolization and regioselective C2-bromination in structurally complex peptides. The TES-mediated approach suppresses the competing reactivity of oxazole, α,β-unsaturated amide, and free amide NH groups that foil direct bromination, and it resolves the long-standing incompatibility between 2-BrTrp and requisite oxidation chemistry. The authors anticipate that this strategy will enable synthetic access to related marine natural products bearing 2-BrTrp motifs, including jasplakinolide and konbamide, and will find broader application in scaffold diversification of medicinally relevant macrocyclic peptides.