Peptides have become increasingly important as drug candidates and research tools due to their favorable safety profiles and ability to modulate protein-protein interactions. However, native peptide therapeutics often suffer from low plasma stability, poor oral bioavailability, and short circulation times in the body. Late-stage chemical modification of amino acid side chains offers a promising strategy to enhance peptide properties without redesigning the entire molecule. Among the 20 common amino acids, tryptophan presents a particularly attractive target for such modifications because of its rarity and its critical roles in molecular recognition.
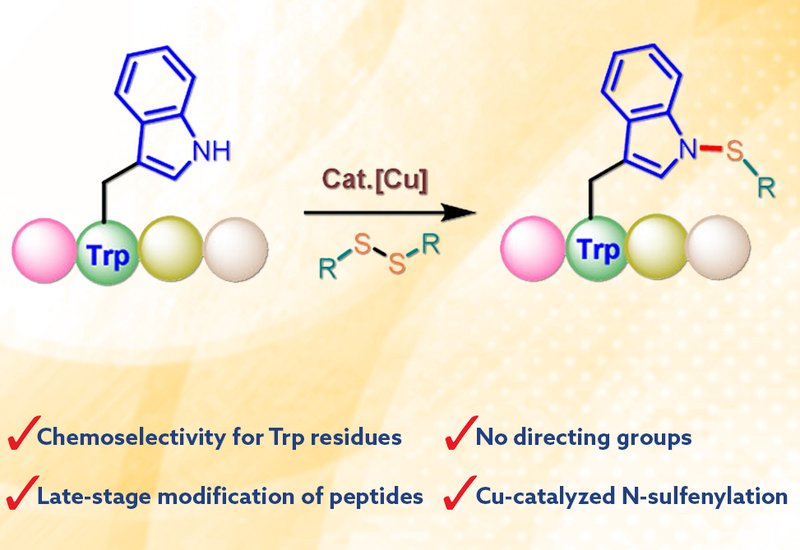
A team of researchers led by Professor Jian Tang at Qufu Normal University, published in Organic Letters, report a copper-catalyzed method for selectively attaching sulfur-containing groups to the nitrogen atom of tryptophan residues in peptides. This N-sulfenylation reaction proceeds efficiently without requiring directing groups or protecting groups on other amino acid side chains, marking a significant advance in peptide functionalization chemistry.
The optimized reaction employs copper bromide as a catalyst with silver carbonate and triethylamine in dichloromethane at 80°C. Control experiments confirmed that each component plays an essential role: removing the copper catalyst, silver additive, or base abolished product formation. The reaction tolerates oxygen and proceeds under ambient air conditions. The team systematically evaluated various dipeptides containing tryptophan paired with different amino acids. Aliphatic residues such as alanine, leucine, and valine reacted smoothly, as did peptides bearing serine, threonine, and tyrosine. Notably, the position of tryptophan within the peptide sequence did not affect reaction efficiency, and both L- and D-tryptophan substrates performed well.
The substrate scope extended to diverse disulfide coupling partners. Diaryl disulfides bearing electron-donating or electron-withdrawing substituents all yielded the corresponding N-sulfenylated products in good yields. Dialkyl disulfides also participated effectively, further broadening the utility of this chemistry. The team then challenged the method with larger peptide substrates. Tripeptides, tetrapeptides, and pentapeptides all underwent selective modification at tryptophan without diminished reactivity as chain length increased.
Mechanistic studies using radical trapping agents suggested that the transformation involves single-electron transfer processes. The silver additive appears to serve dual roles: oxidizing the copper catalyst to generate the active species and coordinating with the disulfide to weaken the S-S bond, facilitating homolytic cleavage to produce sulfide radicals. The proposed catalytic cycle involves copper-mediated radical coupling to form a key intermediate that releases the sulfenylated product through reductive elimination.
The practical utility of this method was demonstrated through several synthetic transformations. The reaction scaled efficiently to gram quantities without loss of yield. The sulfenylated products could undergo further derivatization, including selenylation, olefination via palladium-catalyzed C-H activation, and oxidation to the corresponding sulfone. The N-sulfenyl group could also be removed cleanly using fluoride reagents, providing a route back to the parent tryptophan scaffold if desired.
This chemistry provides a valuable addition to the synthetic toolbox for late-stage peptide modification. The ability to selectively functionalize tryptophan at the nitrogen position opens new possibilities for tuning the physicochemical and pharmacokinetic properties of peptide therapeutics.