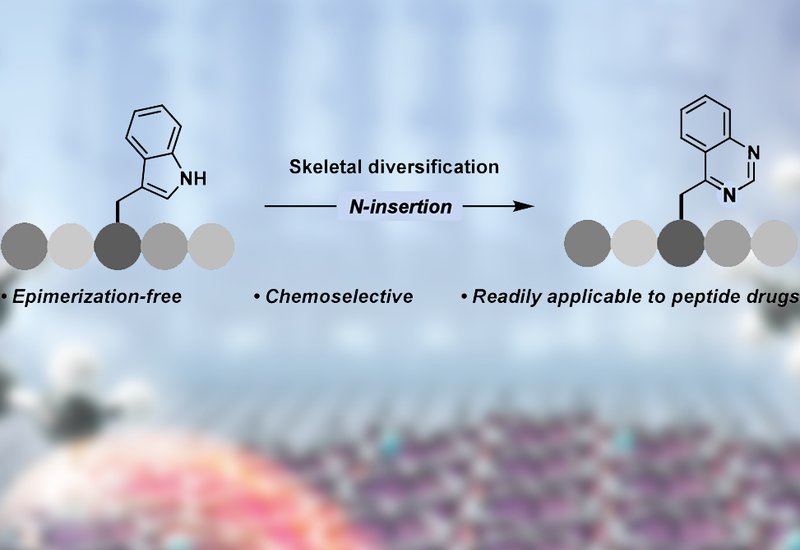
A collaboration between the laboratories of Professors Bill Morandi in the Laboratorium für Organische Chemie and Nathalie Grob in the Institute of Pharmaceutical Sciences, both at ETH Zürich, has developed a method for converting tryptophan's indole ring into a quinazoline scaffold through nitrogen atom insertion, enabling late-stage diversification of peptide drugs without epimerization. Published in the Journal of the American Chemical Society, the work extends atom insertion chemistry to peptide modalities for the first time, successfully modifying therapeutics including the blockbuster GLP-1 receptor agonist semaglutide.
Existing methods for tryptophan modification install functional groups at peripheral positions on the indole ring, leaving the core scaffold unchanged. The Zürich team pursued a fundamentally different strategy: direct skeletal editing that inserts a nitrogen into the five-membered pyrrole portion of indole yielding a six-membered pyrimidine ring. This transformation maintains the planar hydrophobic character valued for protein interactions while converting a hydrogen-bond donor into a scaffold bearing two hydrogen-bond acceptors, potentially providing new handles for modulating drug–target interactions and pharmacokinetics.
The reaction employs ammonium carbamate as a nitrogen source and a hypervalent iodine reagent to generate an electrophilic nitrogen transfer species. Initial conditions developed for simple indoles required strong base, which risked epimerizing peptide stereocenters. The researchers discovered that tryptophan's 3-substitution pattern stabilizes the reaction intermediate sufficiently to proceed without base, and chiral chromatography confirmed complete retention of stereochemistry across L, D, and racemic substrates. The optimized protocol tolerated protecting groups at both termini along with side chains containing aliphatic, hydroxyl, basic, and acidic functionalities. Sulfur-containing residues proved incompatible due to oxidation, and primary amides required rapid quenching to prevent Hofmann rearrangement, but a decision tree guides users through condition selection for challenging substrates.
The method scaled smoothly from dipeptides through pentapeptides, with tryptophan position in the sequence showing no significant effect on yield. Application to approved therapeutics demonstrated practical utility: the gonadotropin-releasing hormone agonist leuprolide converted in 79% yield, the melanocortin receptor agonist bremelanotide in 67%, the antibiotic daptomycin in 20%, and the 31-residue semaglutide in 15% isolated yield. The successful transformation of semaglutide, manufactured commercially by semi-recombinant methods, establishes that nitrogen atom insertion can access structural diversity in medicinally relevant peptides that would otherwise require lengthy de novo synthesis.