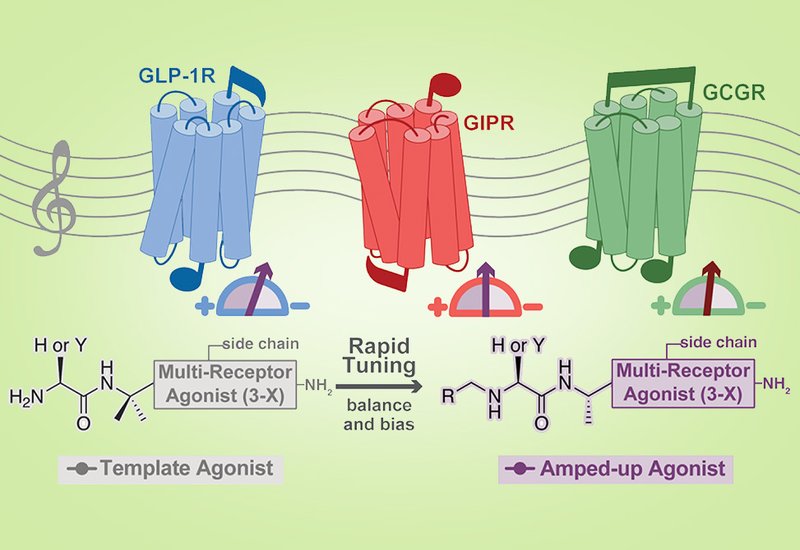
Unimolecular multiagonists that co-activate the glucagon-like peptide-1 receptor, GLP-1R, the glucose-dependent insulinotropic polypeptide receptor, GIPR, and the glucagon receptor, GCGR, have become central to metabolic syndrome therapeutics. Tirzepatide, the first FDA-approved GLP-1R/GIPR dual agonist for type 2 diabetes, and retatrutide, a triple agonist in late-stage clinical development, exemplify the promise of coordinated receptor engagement within a single scaffold. Yet both carry imbalanced receptor profiles. Tirzepatide is roughly 13-fold less potent at GLP-1R than native GLP-1 while reaching full GIPR efficacy, and retatrutide is roughly 2.5-fold less potent at GLP-1R while achieving 9-fold higher maximal activity at GIPR, where strong engagement is anti-emetic. The chemical logic for systematically adjusting that balance remains poorly defined. Small sequence changes propagate nonlinearly across receptors, and meaningful retuning has typically required wholesale scaffold redesign rather than targeted, modular edits. The field has lacked a compact handle for incremental, interpretable control of receptor weighting and pathway preference.

On the cover, the three incretin receptors, GLP-1R, GIPR, and GCGR, become input channels on a mixing board. Knobs for potency, bias, and balance shape the signal, and N-terminal chemistry is the hand on the dials. And, of course, all dials go to eleven.
Researchers in the Kumar Group and the Beinborn Group at Tufts University, published in J. Med. Chem., addressed this gap by exploiting the peptide N-terminus as a precision lever. Because the N-terminus of class B GPCR ligands reaches deep into the membrane-embedded orthosteric pocket, localized chemistry at that site can redirect receptor activation trajectories without perturbing the rest of the scaffold. The team built matched dual- and triagonist templates derived from previously reported balanced scaffolds and screened four categories of N-terminal edits: reductive-amination alkyl capping, electrophilic fluoroalkylation via iodonium reagents, substitution of the first residue with non-canonical amino acids, and combination constructs pairing backbone residue substitution with N-alkylation. All modified peptides were assembled by fast-flow Fmoc solid-phase synthesis and characterized by RP-HPLC and ESI-MS. Receptor potency was quantified with a cAMP-dependent luciferase reporter assay, and β-arrestin-2 recruitment was measured by bioluminescence resonance energy transfer, BRET.
On monoagonist templates, N-terminal chemistry proved strongly directive in a chemotype-dependent manner. A p-hydroxybenzyl appendage on GLP-1 raised GIPR potency from an EC50 of 1 μM to 30 nM while preserving GLP-1R activity; pairing that group with a His7Tyr substitution yielded 9-GLP1(H7Y), the most potent GLP-1-based GIPR ligand in the series, with an EC50 of 10 nM at GIPR. On the GIP scaffold, a Tyr1His substitution combined with N-methylimidazole alkylation conferred measurable GLP-1R activity at 0.1 μM, a roughly 30-fold gain relative to native GIP. By contrast, trifluoroethylation at the same position left receptor selectivity unchanged, establishing it as a balance-neutral protease-protection strategy. On the GLP-1R/GIPR dual-agonist template, p-hydroxybenzylation shifted the potency ratio to favor GIPR 19-fold over GLP-1R, whereas methyl-imidazole alkylation combined with the Tyr1His substitution achieved the reverse, maintaining near-picomolar GLP-1R potency at 14.5 pM while weakening GIPR engagement to 57 pM.
The triagonist series extended these principles to a three-receptor system and revealed additional axes of control. An adamantyl appendage preserved GLP-1R potency at 13.4 pM while dropping GIPR and GCGR potencies to 287 pM and 573 pM, respectively. A p-trifluoromethylphenylalanine substitution at position 1 left GLP-1R potency nearly unchanged but reduced GIPR and GCGR potencies by roughly 23-fold and 45-fold, demonstrating that aromatic electronics at residue 1 can decouple GLP-1R engagement from the other two receptors. Combining a His1Tyr substitution with p-hydroxybenzyl alkylation produced 9-TA(H1Y), which favored GIPR with only a modest 3-fold loss relative to the parent triagonist, while GLP-1R and GCGR potency fell roughly 40-fold and 12-fold, a profile reminiscent of retatrutide. A paired His1Tyr and Thr7Ile double substitution selectively suppressed GLP-1R activation to 169 pM without materially altering GIPR or GCGR potency. On the signaling-bias axis, trifluoroethylation of the triagonist N-terminus raised β-arrestin-2 maximal recruitment to 100% of the GLP-1 reference, whereas the same trifluoroethyl group combined with Thr7Ile, in 2-TA(T7I), collapsed β-arrestin-2 recruitment to near-basal levels while preserving cAMP potency, inverting bias with a single neighboring amino-acid change. N-terminally alkylated multiagonists, including 2-DA, 11-DA(Y1H), 7-TA, 9-TA, and 11-TA(H1Y), also resisted DPP4-catalyzed degradation in cell-based protease assays, confirming that receptor tuning and protease protection can be layered concurrently.
These results establish N-terminal chemistry as a modular, scaffold-preserving grammar for programming incretin multiagonist pharmacology. The ability to shift receptor potency ratios by orders of magnitude, tune cAMP-vs.-β-arrestin-2 signaling balance, and confer DPP4 resistance through a single site opens a route to systematic exploration of receptor set-points that remain undefined for optimal glycemic control, weight loss, and tolerability. As the polypharmacology field expands to incorporate amylin, PYY, FGF21, and other receptors alongside GLP-1R, GIPR, and GCGR, the compact N-terminal editing strategy described here offers a transferable platform for building next-generation unimolecular agonists with precisely dialed activity profiles.