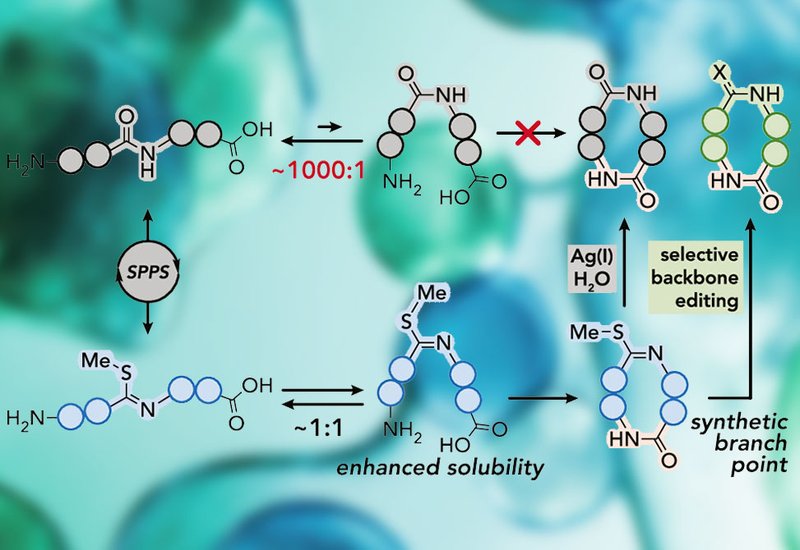
Head-to-tail macrocyclic peptides occupy a coveted niche in drug discovery. Lacking free termini, they resist exoprotease degradation, and their compact conformations shield backbone amides, promoting passive membrane diffusion and sometimes oral bioavailability. Yet for sequences of four to five residues, especially homochiral all-L sequences, the chemistry is brutally unforgiving. Peptide backbones overwhelmingly prefer the trans-amide geometry, above 99.8% per amide bond, which holds the chain in an extended conformation that resists entropically costly ring closure. The result is oligomers, poor yields, and frequent outright failure. Side-chain-protected short peptides also aggregate and dissolve poorly in organic solvents, sometimes making cyclization impossible to attempt at useful concentrations.
Researchers in the VanVeller Group at Iowa State University, published in J. Am. Chem. Soc., hypothesized that thioimidates, backbone-localized functional groups that maintain a near 1:1 dynamic equilibrium between cis and trans isomers, could solve both preorganization and solubility at once. Variable-temperature NMR confirmed that interconversion proceeds on the millisecond timescale at room temperature, with a barrier of 14 kcal/mol and a free energy difference of only 0.25 kcal/mol. The team compared thioimidate peptides against all-amide controls across tetra- and pentapeptide sequences at 5, 10, and 15 mM using pentafluorophenyl diphenylphosphinate as the coupling reagent.
The solubility advantage proved dramatic. Thioimidate peptides dissolved above 100 mM in DMF and DCM, while all-amide controls required a solubility-enhancing Gln(Trt) residue just to reach 5 mM. Cyclization yields tracked the same pattern. A homochiral tetrapeptide thioimidate closed in 10 to 12% yield where the all-amide control produced nothing. Adding a C-terminal D-Ala, a standard rescue strategy, failed to help the all-amide version but pushed the thioimidate analogue to 62 to 70%. A homochiral pentapeptide thioimidate cycled at 43 to 50% yield even at 50 mM, a concentration where the all-amide control remained insoluble. A mild Ag(I) step then converts the thioimidate to the native amide, with AgBF4 in 1:9 H2O/1,4-dioxane giving 84% conversion. Applied to seven intentionally difficult homochiral sequences, the strategy delivered several previously inaccessible pentapeptide macrocycles, the first synthesis of the trichoderide A tetrapeptide core from Trichoderma reesei, and the first synthesis of the Streptomyces cyclotetrapeptide vinaceuline. A homochiral cyclopentaleucine macrocycle previously isolated in 3% yield was obtained in 34% yield without optimization.
Beyond enabling ring closure, the thioimidate serves as a traceless branch point for late-stage backbone editing. From a single cyclic precursor, the team accessed four structurally distinct analogs: a 15N-labeled amidine prepared with inexpensive 15NH4OAc, a thioamide produced with 2,4,6-collidine and H2S in 66% yield, a deuterium-labeled reduced-amide pseudopeptide generated in 94% yield by NaCNBD3 reduction, and an 18O-labeled native amide obtained in greater than 99% yield by running the Ag(I) hydrolysis in H218O. The reduction protocol is the first general amide-bond reduction of a cyclic peptide without ring opening, and the 18O route is the first selective chemical method for amide-oxygen labeling compatible with standard SPPS protecting groups.
The thioimidate platform fills a longstanding gap in macrocyclic peptide synthesis. It is side-chain-agnostic, works with conventional SPPS protecting-group logic and coupling reagents, and leaves no permanent trace in the final macrocycle. Medicinal chemists gain a practical route to thioamides, amidines, pseudopeptides, and isotopically labeled derivatives from one shared intermediate, enabling systematic exploration of backbone bioisosteres. Trityl-protected cysteine is incompatible with the Ag(I) step, though methionine tolerates the conditions, suggesting that alternative cysteine protecting groups could extend future scope. With first syntheses of two natural products already demonstrated, the platform looks set to accelerate both the discovery of biologically active macrocycles and the study of how backbone chemistry governs peptide conformation and cell permeability.