Recent Peptide Publications

Intracellular Peptide Stapling
Mason Lab
Mar 13, 2026
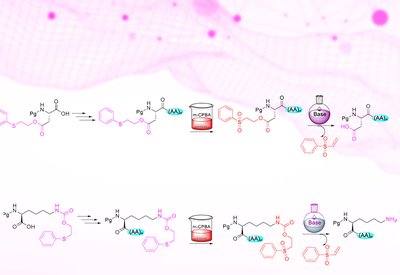
Safety Catch Strategy
Albericio Lab
Mar 12, 2026
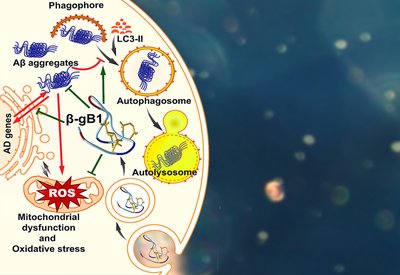
Ginkgo Degrader
Tam Lab
Mar 11, 2026
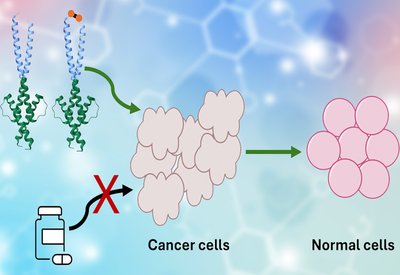
Frankenproteins Block Myc
Shin Lab
Mar 10, 2026
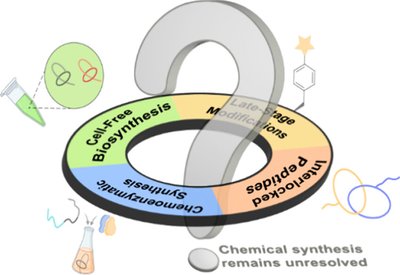
Lasso Peptide Progress
Craik Lab
Mar 9, 2026
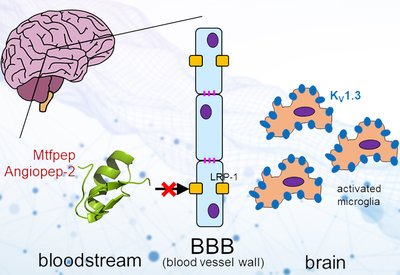
Barrier Shuttle Peptides
Norton Lab
Mar 6, 2026

Targeted Fusion Inhibitors
Cheloha Lab
Mar 6, 2026
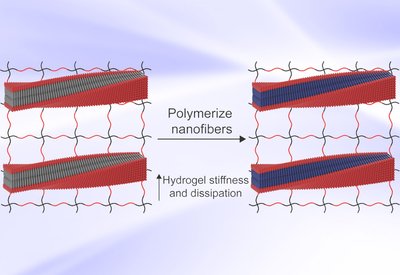
Programmable Hydrogel Mechanics
Pashuck Lab
Mar 6, 2026
Hemostatic Protein Polymers
Nash Group
Mar 6, 2026
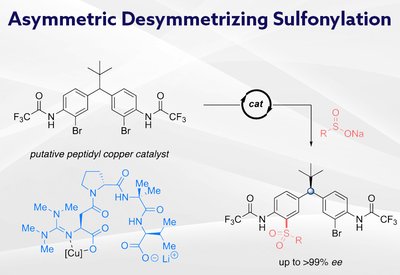
Remote Stereocontrol
Miller Lab
Feb 26, 2026
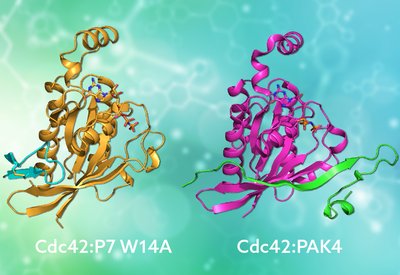
Mimicking Nature's Grip
Mott and Owen Lab
Feb 20, 2026
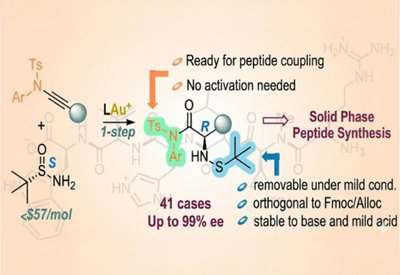
Ready-Made Amino Acids
Liming Zhang Lab
Feb 18, 2026
Peptide Pioneers
Celebrating new faculty launching independent peptide research.
Andrew Brennan
Assistant Professor
Brennan Lab, University of Bath
Andy Brennan's path through peptide science reads like a tour of the UK's leading protein chemistry groups, each stop adding new tools to his repertoire. His introduction to peptides began during his undergraduate studies at the University of Bristol in the laboratory of Dek Woolfson, where he explored coiled-coil helical …
Read More
Peptide Postdocs
Recognizing postdoctoral researchers advancing peptide science.
Danielle Morgan
Postdoctoral Research Fellow
MacMillan Group, Princeton University
Danielle Morgan had just submitted her Ph.D. thesis at the University of Glasgow when she boarded a flight to Whistler for the 27th American Peptide Symposium in June 2022. The airline lost her luggage. With a flash talk scheduled for the first morning, she dashed to local shops and found …
Read More
Peptide Primers
Educational resources for peptide science.
Peptide Synthesis for Beginners
3 sections
Learn the fundamentals of solid-phase peptide synthesis (SPPS) with this comprehensive guide. Covers equipment setup, Fmoc chemistry, coupling reactions, cleavage protocols, and HPLC purification.
Start Learning
Student Spotlight
Highlighting outstanding graduate students shaping the future of peptide science.
Arina Filatova
Undergraduate Student Researcher
Hackeng and Dijkgraaf Groups, Maastricht University
Arina Filatova is a BSc student in Chemical Biology at Maastricht University. Her fascination with biological macromolecules began early in her studies, when she first learned that a protein's three-dimensional structure dictates its function. This curiosity evolved into a central academic interest in how chemical structure governs biological activity...
Read More
Global Peptide Groups
Featuring Internationally Notable Peptide Science Research Groups.
The ChemSynBio Group
Benjamí Oller-Salvia · IQS School of Engineering, Ramon Llull University, Barcelona, Spain
When Benjamí Oller-Salvia returned to Barcelona in 2019, he had a faculty position and no funding. Six years later, his ChemSynBio group at IQS School of Engineering has grown to 20 researchers from nine countries, united by a shared challenge: getting therapeutics past the blood-brain barrier. From venom-inspired shuttle peptides …
Read More
Peptide News
The latest from the peptide science community.

Summer School in Peptide Science
The Australian Research Council Centre of Excellence for Innovations in Peptide and Protein Science, CIPPS, has announced the inaugural Summer School in Peptide Science, SSIPS. The Summer School will run in conjunction with the 5th International Conference on Cyclic Peptides, ICCP, at Heron Island Resort on the Great Barrier Reef, Australia, from 12–19 July 2026.
The program is designed for the training of graduate students and early-career postdoctoral researchers. The faculty comprises leading international experts who will deliver lectures and workshops spanning peptide discovery, green chemistry, computational design, and peptide translation.
Mar 1, 2026
Read More
Call for Nominations
The 2027 R. Bruce Merrifield Award recognizing the lifetime achievement of a peptide scientist, whose work exemplifies the highest level of scientific creativity will be presented at the 30th American Peptide Symposium in Boston, MA, June 20-24, 2027.
Nominating documents should be submitted by April 15th, 2026.
Feb 19, 2026
Read More
Cooperativity Unlocked - The Team
Each name appearing in the authors' line of a research article carries a story. The Biochemistry paper from the Waters laboratory at the University of North Carolina, which revealed how cooperative binding interactions govern selectivity in histone reader proteins, is no exception. Behind the science stands a team whose trajectories from Chapel Hill now span academia and industry, from North Carolina to South San Francisco.
Feb 18, 2026
Read More
Partners in Discovery
When Michael Bertucci and Yftah Tal-Gan set up their posters side by side at the 2015 American Peptide Symposium in Orlando, they discovered something remarkable: both young assistant professors had independently chosen to study quorum sensing in streptococci.
That serendipitous encounter sparked a decade-long collaboration spanning Lafayette College and the University of Nevada, Reno. Their latest work in ACS Infectious Diseases, highlighted on our website, reveals how Streptococcus gordonii uses peptide signaling to produce hydrogen peroxide, a natural weapon against cavity-causing bacteria. Just as bacteria coordinate through chemical crosstalk, these research allies have built a partnership that bridges coasts, institutions, and career stages.
Feb 2, 2026
Read More